
Effect of Exchange–correlation Functionals on Ground State Geometries, Optoelectronic and Charge Transfer of Triphenylamine-based Dyes
Published 2022-04-08
Keywords
- Charge transfer; DFT; optoelectronic; triphenylamine; XC functional
How to Cite
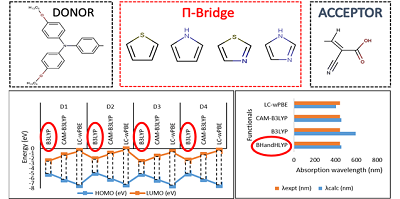
Abstract
The importance of the Density Functional Theory (DFT) calculation approach lies in their ability to provide a highly accurate prediction of structural and optoelectronic properties. However, the traditional methods of DFT failed to predict optoelectronic properties satisfactorily. Therefore, it will be necessary to examine methods containing different percentages of Hartree-Fock exchange and correlation in order to find the most suitable functionals. DFT and Time-Dependent-DFT (TD-DFT) calculations was carried out using four different functionals approximations incorporating a different amount of Hartree Fock exchange (B3LYP, BHandHLYP, CAM-B3LYP and LCωPBE), in order to evaluate their accuracies to predict the geometrical, optoelectronic and charge transfer properties of four triphenylamine-based dyes for Dye-Sensitized Solar Cells (DSSCs) applications. The functional hybrid B3LYP was the best among adopted functional that reproduced the geometrical, optoelectronic and charge transfer properties. On the other hand, it has been shown that the Hartree-Fock exchange percentage for BHandHLYP, significantly improved TD-DFT results in the case of organic dyes. Moreover, the corrected long-range functionals (CAM-B3LYP and LC-wPBE) present valuable tools for giving results of comparable precision with experimental optical data. In terms of the choice of the most appropriate functional for computational calculation, the obtained results can be useful for future DSSC applications.