
Bilastine Based Drugs as SARS-CoV-2 Protease Inhibitors: Molecular Docking, Dynamics, and ADMET Related Studies
Published 2022-04-07
Keywords
- Inhibition constant; Drug-likeness; Molecular docking; Molecular Dynamics; SARS-CoV-2
How to Cite
Abstract
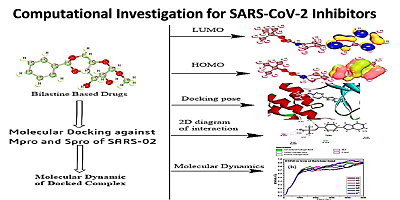
Bilastine drugs, structurally piperidine-1-carboxylate and sulfonyloxyethyl carboxylate derivatives, have significantly been employed as the medication of second-generation antihistamine drugs, and are used for the treatment of allergic rhinoconjunctivities and urticarial (hives). The bilastine drugs, composed of benzene carboxylate, propanoate, carboxylate, methyl-sulfonate, propanoic acid, butanoic acid, and pentanoic acid derivatives, were investigated through computational tools against SARS-CoV-2. The COVID-19 virus consists of five proteases where the curial function is performed by main proteases (Mpro) and Spike proteases (Spro). The Mpro and Spro were selected for calculation of molecular docking by these bilastine drugs which showed higher binding energy (<-6.5 kcal/mol) for both proteases. The main carboxylic acid group in bilastine drugs is found the primary key for a high binding score to show the large binding affinity with Mpro and Spro, and is highly responsible for forming the hydrogen bond although the various hydrophobic bonds were produced as a weak interaction. For justification, the stability of molecular docked ligand-protein complexes was investigated with molecular dynamics. It showed that the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) of all these drugs were below the 0.9 Å after residue interaction. Moreover, the HOMO-LUMO gap, hardness, and softness provided full details for their chemical reactivity. In this view, the pharmacokinetics and Lipinski rule were calculated, and all of these molecules had satisfied the Lipinski rule. Finally, using the admetSAR online database, absorption, distribution, metabolism, excretion, and toxicity have been calculated which indicated that these bilastine drugs are non-carcinogenic and less harmful for both aquatic and non-aquatic species.