
Published 2021-07-06
Keywords
- DFT Calculations,
- Electronic Structure,
- Perovskite,
- Strontium Stannate (SrSnO3)
How to Cite
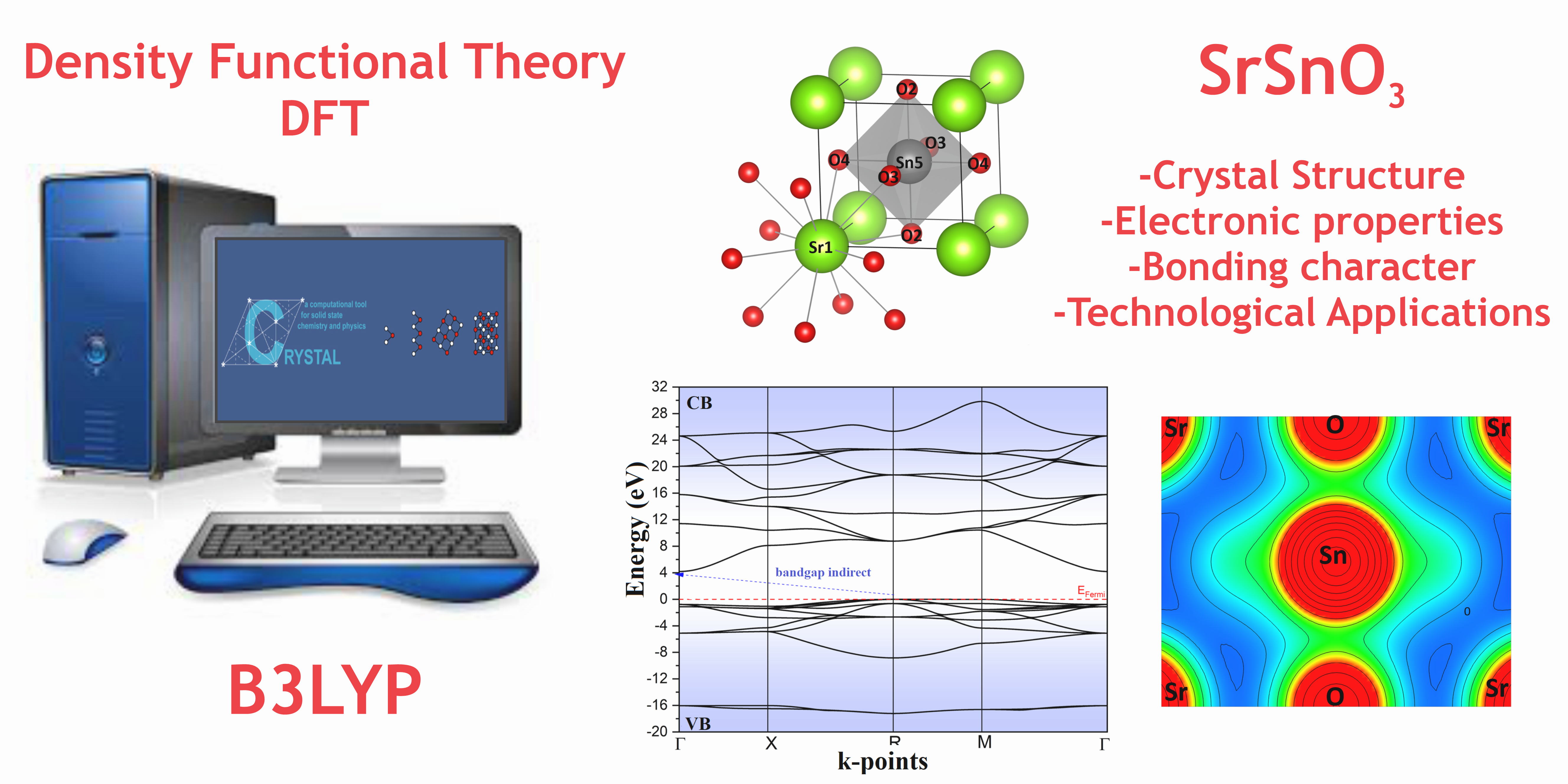
Abstract
This article reports a theoretical study on structural and electronic properties of the cubic strontium stannate (SrSnO3) using periodic quantum-mechanics calculations within the Density Functional Theory method combined with B3LYP exchange-correlation functional, as implemented in the CRYSTAL14 code. The results were analyzed using the energy level diagram, atomic orbital distributions, and electron density maps. The structural analysis confirmed the SrSnO3 cubic symmetry, and the electronic properties were associated with [SrO12] and [SnO6] clusters with distinct bonding character. Furthermore, our structural and electronic calculations are in good agreement with the available experimental data showing a mean percentage error close to 2.2% for the structural parameter and paving the avenue towards the complete understanding of the overall properties of perovskite materials.