
Structural Elucidation, DFT Calculations, Hirshfeld Surface Analysis, Molecular Dynamics Simulation, ADMET Profiles, and Molecular Docking of Two Benzazocine Derivatives NAOP-12 and NEMKH-12
Published 2025-04-27
Keywords
- Molecular Dynamics,
- Molecular Docking,
- ADMET,
- DFT,
- Fukui functions
- Hirshfeld surface ...More
How to Cite




Abstract
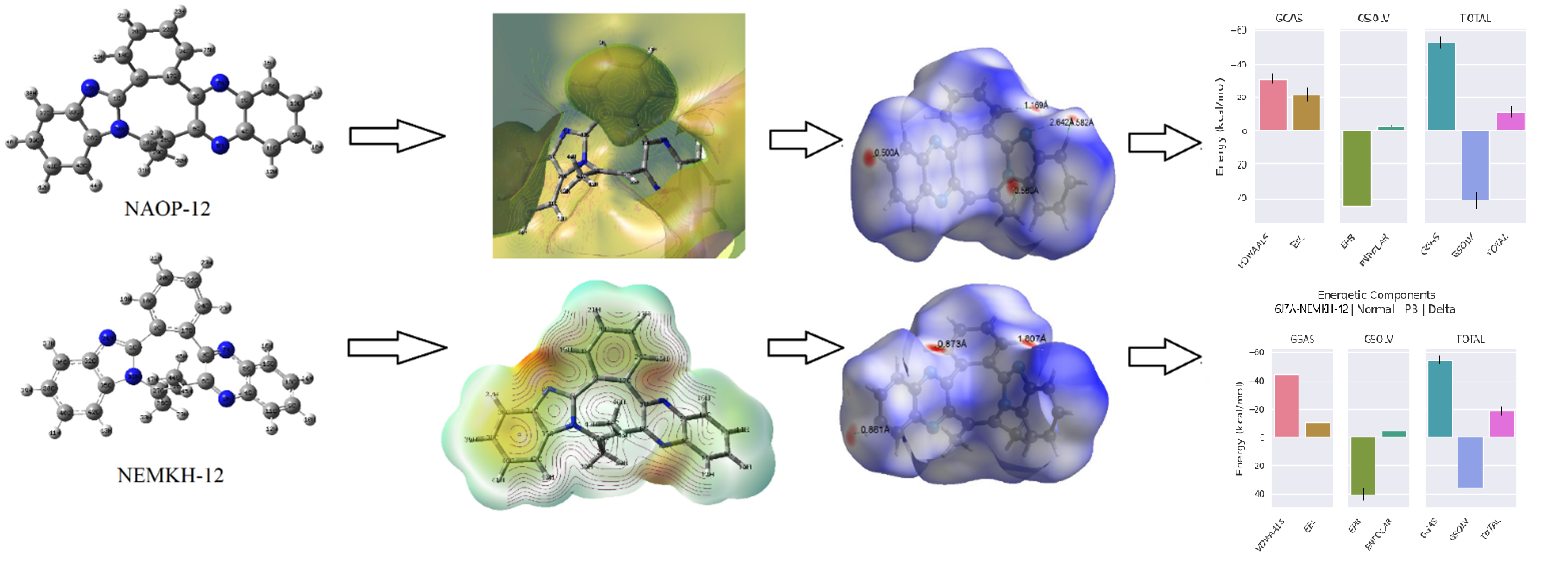
In this study two novel benzazocine derivatives containing potentially anticancer properties, (a) NAOP-12 (C24H16N4) & (b) NEMKH-12 (C25H18N4), were investigated utilizing quantum computational technique. The equilibrium geometries of the compounds have been obtained and analyzed using the DFT-B3LYP/6-311++G(d,p) method. FT-IR analysis was used to identify the various functional groups, and the results are compared to the simulated spectra. The oscillation modes were examined theoretically. The electronic properties such as HOMO and LUMO energies and associated frontier energy band gap were calculated. Molecular Electrostatic Potential predictions are done to analyze the electrophilic and nucleophilic centers. Electrical parameters like dipole moment, molecular polarizability and first static hyperpolarizability, have been utilized to predict the biological nature and non-linear optical (NLO) behavior of the molecules. Chemically active sites are described using Fukui functions, chemical softness, and other descriptors. The Hirshfeld surface analysis has been performed to investigate the weak interactions in the molecules. A 500 ns molecular dynamics simulation was conducted, evaluating models using RMSD, RMSF, Rg, SASA metrics and the gmxMMPBSA tool was utilized for free energy calculations. The molecular docking analysis has been performed with the fusion proteins 6J7A and 6J7I. The drug-likeness and ADMET parameters were computed to investigate their pharmaceutical behavior.